
 About ALPHANATE
About ALPHANATE
ALPHANATE provides natural protection through its FVIII/VWF complex
About ALPHANATE: A hemophilia A and von Willebrand disease treatment
ALPHANATE is a plasma-derived factor complex containing both factor VIII (FVIII) and von Willebrand factor (VWF). The natural FVIII/VWF complex in ALPHANATE is carefully preserved during the production process.1
ALPHANATE provides proven protection for both adult and pediatric patients with hemophilia A or with von Willebrand disease (VWD) undergoing surgical and/or invasive procedures.1
ALPHANATE is contraindicated in in patients who have manifested life-threatening immediate hypersensitivity reactions, including anaphylaxis, to the product or its components
Role of FVIII/VWF complex
The naturally occurring FVIII/VWF complex in ALPHANATE plays a critical role in hemostasis.
VWF binds, carries, and protects FVIII2
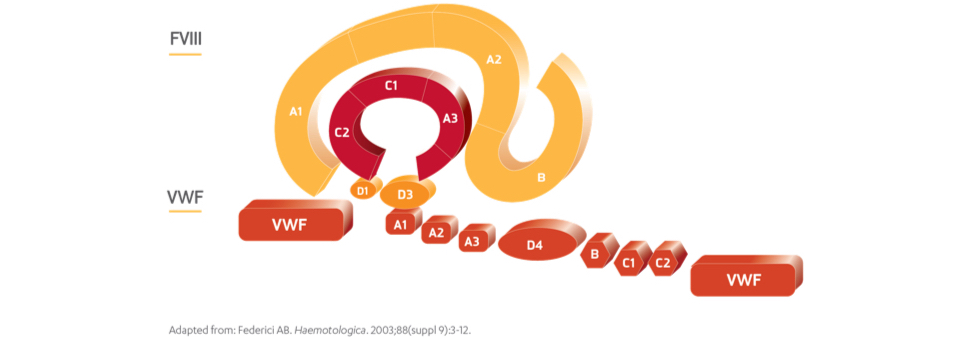
- VWF binds to FVIII and thereby protects it against attack by a variety of proteases3,4
- VWF carries FVIII to the bleeding site where FVIII is cleaved and activated by thrombin5
- VWF prolongs the half-life of circulating FVIII3
- VWF blocks endocytosis and subsequent presentation of FVIII peptides to dendritic cells4
- VWF masks antibody epitopes on the light chain of FVIII involved in inhibitor development4
Bleeding disorder treatment goals can be met using a plasma-derived product that contains both FVIII and VWF.
Indication
ALPHANATE® (antihemophilic factor/von Willebrand factor complex [human]) is indicated for:
- Control and prevention of bleeding episodes and perioperative management in adult and pediatric patients with factor VIII (FVIII) deficiency due to hemophilia A.
- Surgical and/or invasive procedures in adult and pediatric patients with von Willebrand disease (VWD) in whom desmopressin (DDAVP) is either ineffective or contraindicated. It is not indicated for patients with severe VWD (type 3) undergoing major surgery.
Important Safety Information
ALPHANATE is contraindicated in patients who have manifested life-threatening immediate hypersensitivity reactions, including anaphylaxis, to the product or its components.
Anaphylaxis and severe hypersensitivity reactions are possible with ALPHANATE. Discontinue use of ALPHANATE if hypersensitivity symptoms occur, and initiate appropriate treatment.
Development of procoagulant activity-neutralizing antibodies (inhibitors) has been detected in patients receiving FVIII-containing products. Carefully monitor patients treated with AHF products for the development of FVIII inhibitors by appropriate clinical observations and laboratory tests.
Thromboembolic events have been reported with AHF/VWF complex (human) in VWD patients, especially in the setting of known risk factors.
Intravascular hemolysis may occur with infusion of large doses of AHF/VWF complex (human).
Rapid administration of a FVIII concentrate may result in vasomotor reactions.
Because ALPHANATE is made from human plasma, it may carry a risk of transmitting infectious agents, eg, viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent, despite steps designed to reduce this risk.
Monitor for development of FVIII and VWF inhibitors. Perform appropriate assays to determine if FVIII and/or VWF inhibitor(s) are present if bleeding is not controlled with expected dose of ALPHANATE.
The most frequent adverse drug reactions reported with ALPHANATE in >1% of infusions were pruritus, headache, back pain, paresthesia, respiratory distress, facial edema, pain, rash, and chills.
Please see full Prescribing Information for ALPHANATE.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch or call 1.800.FDA.1088.
References:
- ALPHANATE® (antihemophilic factor/von Willebrand factor complex [human]) Prescribing Information. Grifols.
- Federici AB. Haematologica. 2003;88(suppl 9):3-12.
- Federici AB, Mannucci PM. Ann Med. 2007;39(5):346-358.
- Franchini M, Lippi G. Thromb Haemost. 2010;104:931-940.
- Lacroix-Desmazes S, et al. Blood. 2008;112(2):240-249.